The goal of this week's lab is to:
We will be doing the following problems in-lab. Given the matrix in your labs/3/input directory titled nj_inclass.dist:
Neighbor Joining is the most popular distance-based method for inferring tree structures given its ability to handle non-ultrametric data. You will write a program NJTree.py with the following objective:
An example run for your program would be:
$ ./NJTree.py distancesWhere distances is the name of the file containing the distance matrix.
Your distance file will contain one line per pairwise distance in the format:
A B d_A,Bwhere A and B are two different taxa (e.g., genes, organisms) and d_A,B is a positive valued float.
To name your clusters, simply merge the names of the two sub-clusters with a hyphen in between, e.g. merging A and B yields a parent cluster A-B. Merging parent cluster A-B with C-D would yield A-B-C-D (the ordering of letters does not matter).
Your program should output a list of edges in your tree in a similar format to the input, but using distances from the resulting tree as well as including inferred parent clusters:
A A-B d_A,A-B B A-B d_B,A-BYou should only write one line per edge; that is, there should be no distance between A and B since they are not directly connected by an edge.
Your program should output a visualization of your tree using the GraphViz package. This is a popular tool used throughout engineering and science disciplines to draw graphs. Python has a library that makes integrating GraphViz into your program fairly easy.
To use in your program, first import the Python GraphViz library:
import pygraphviz as pgvNOTE: recall that a tree is a special type of graph, so you should use the graph object in GraphViz to represent your tree. To create a graph T and add nodes and edges:
T = pgv.AGraph() #Constructs a graph object
T.add_node("A") #The string is both the label and hash key
T.add_node("B")
T.add_edge("A","B",label="1.0", len=1.0) #Takes in two node keys, an edge label, and the length of the edge
Your tree should have exactly one node for each sequence in the set, and then add one additional node for every parent cluster (i.e., on every merge). The program should have no edges initially, but will add two edges for every merge operation (an edge from each child to the new parent cluster). The edge label and len should be the distance between the child and parent; i.e., d_ik between the two clusters i and k. I suggest setting the len field to a minimum value of 1.0 to prevent overlapping nodes.
Lastly, you will output your graph to a file by using the draw command:
T.draw("tree.png",prog="neato") #neato is the algorithm for drawing the graph
You can use your favorite image viewer to view your results.
Using the in-lab example:
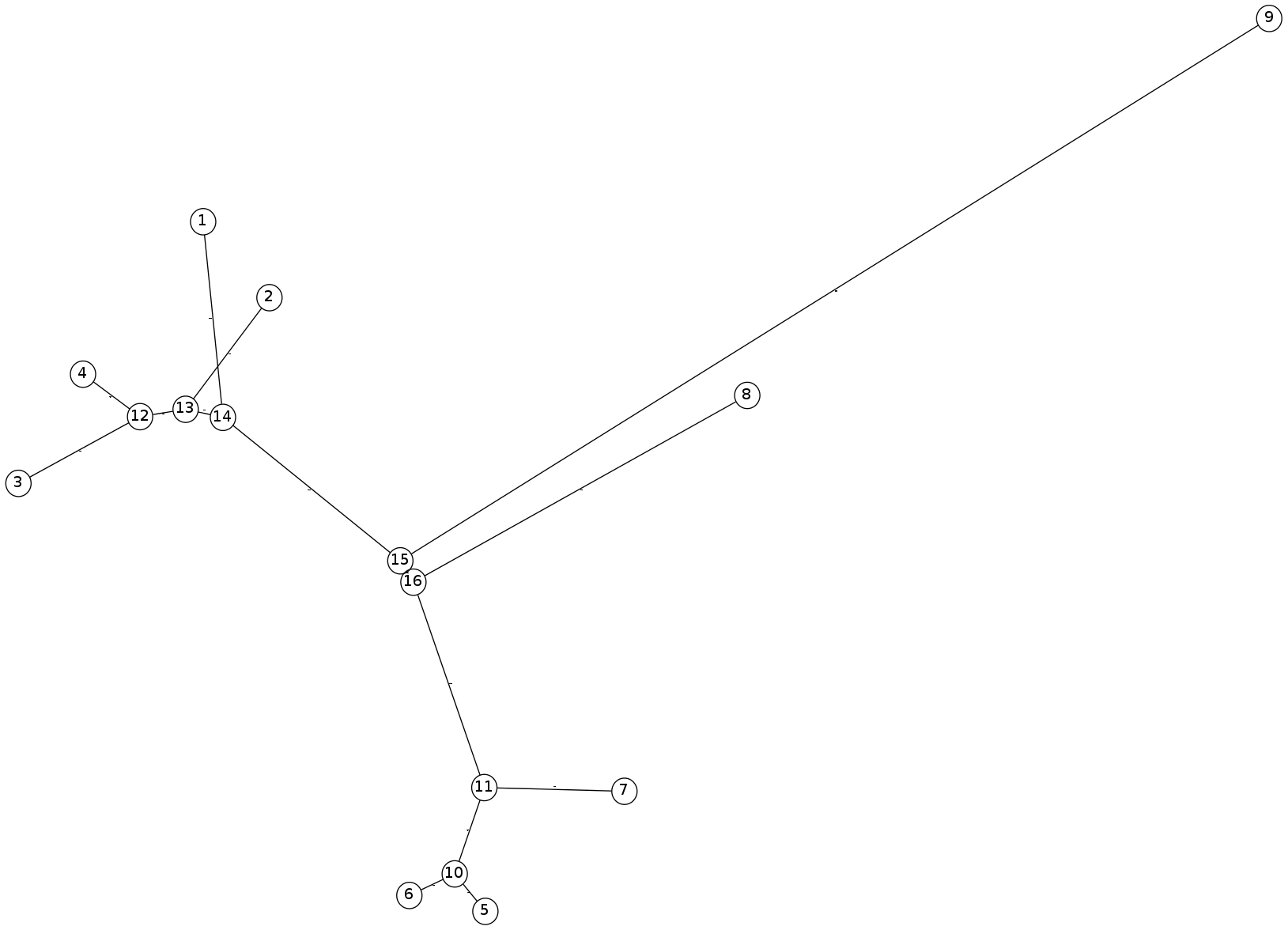
$ ./NJTree.py input/nj_inclass.dist A A-B 6.0 B A-B 1.0 A-B A-B-C 3.0 C A-B-C 2.0 D A-B-C 5.0The resulting image: In-class Example
You should see an input file primate.dist in your input directory. Run your program on this matrix and submit the resulting png as primate.png in your lab directory These distances are scaled distances of mitochondrial DNA found in several primate organisms. I have abbreviated the names to improve readability of the graph image (the nodes become very large with long labels). The mapping is as follows:
For the next set of questions, take a look at the following image produced by the neighbor joining algorithm for studying HIV. The numbers in the nodes map to various strains of HIV (human immunodeficiency virus) and SIV (simian immunodeficiency virus). You can take a look at the mapping and original distance matrix.
AIDS (Acquired Immune Deficiency Syndrome) is caused by two variants of HIV in humans - HIV-1 and HIV-2. You will use the tree to answer questions about the evolution of these two variants, in particular focusing on their relationship with various strains of SIV.
For the second part of your lab, you will approach tree inference using parimony. In a file called weightedParsimony.py, write a program with the following objective:
$ ./weightedParsimony.py tree substitution_matrix leaf_sequences
Leaf Sequences: Contains a list of all of the K sequences representing the leaves of the tree. Each line will be in the format:
taxa_id: sequencewhere taxa_id is the name of the organism or gene, and shoulod match the a leaf in the tree input file, and sequence is a DNA sequence. Using the example we have from class, I have labeled each leaf sequence as L1 through L4 in input/wp_inclass.seq
Tree File: The tree file will define the topology of the phylogenetic tree you are attempting to score. The first line specifies the root. Each line after that is in the format:
child parentFor example, the simple tree from class (input/wp_inclass.tree) would be:
N1 N2 N1 N3 N1 L1 N2 L2 N2 L3 N3 L4 N3Note that the root has been labeled N1. It has two children N2,N3. The leaves connect to either N2 or N3.
Weighted Cost Matrix: Specifies the substitution value for each pair of states possible at each position in the sequence. This has the exact same format as the distance matrix in the Neighbor Joining algorithm; that is, each line contains a pair of characters followed by their substitution score. You can find the example from class in input/wp_inclass.cost. The diagnol is not defined, but you should set it to 0 when you create your substitution function.
Your program should print the total tree score as well as the inferred state of all common ancestors after running the weighted parimony algorithm. Each line should be specified as:
identifier: sequencesimilar to the input sequence file. The in-lecture example would appear as follows:
$./weightedParsimony.py wp_inclass.tree wp_inclass.cost wp_inclass.seq Score: 9.0 Inferred States: L4: G L2: C L3: T L1: A N1: T N2: T N3: TFor debugging purposes, you should output intermediate calculations. You can see my intermediate outputs here. When submitting, please remove this intermediate prints or ensure the required output appears at the end of the file.
While you can construct a tree data structure to help implement your program, it is probably much easier to just use a dictionary instead. For example you can have a dictionary where the key is the identifier (e.g., "N1") and the value is a list of the children (e.g., ["N2","N3"]). A leaf node simply has an empty list for its value. If you need to represent parents, you can have a companion dictionary where the keys and values are reversed.
You should first get your program to work on single-character sequences as practiced in class. Once that is complete, you will want to improve your algorithm to calculate the cumulative score and trace for a set of sequences of length N. You can assume that each leaf sequence is of the same length. Also, since we assume independence of positions, you should be able to just place a loop around the function you already created (you are practicing modularity, right?). I have given a second set of example files beginning with the prefix mammals. A sample run will output:
$./weightedParsimony.py mammals.tree mammals.cost mammals.seq Score: 11.0 Inferred States: Aardvark: CAGGTA Chimp: CGGGTA Dog: TGCACT Bison: CAGACA Elephant: TGCGTA C3: CGGGTA C2: TGCGTA C1: CAGGTA C4: CGCGTAAn output of intermediate values can be found here.
When you are finished with your lab, submit your code using handin68. You should submit, at a minimum:
{kind=link}
{kind=link}